
Science moves forward when researchers verify their and others’ results.
“Reproducibility in scientific research is a prominent issue, and molecular simulations, which play an important role in many subfields of science and engineering, pose particular computational challenges,” said Peter Cummings, associate dean for research and John R. Hall Professor of Chemical Engineering.

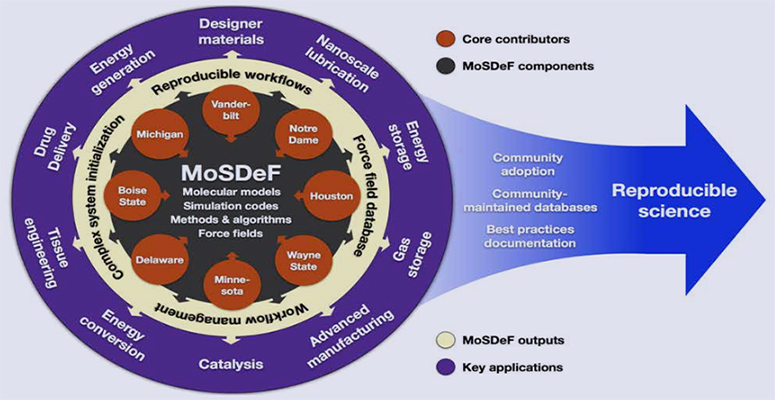
To address the challenges, Cummings, Clare McCabe and their colleagues in the Multiscale Modeling and Simulation group and computer scientists, particularly Akos Ledeczi, in Vanderbilt’s Institute for Software Integrated Systems, developed a robust suite of open-source software tools. The Molecular Simulation and Design Framework (MoSDeF) expedites reproducible computer simulations.
Reproducibility is an essential part of the scientific method. But a crisis of reproducibility, and hence confidence, gained currency over the last decade as disappointing results emerged from large-scale projects to reproduce studies in some medical and science fields.
The ability to close the reproducibility gap has important stakeholders and widespread interest. A $3 million NSF grant has provided support for the Cummings, McCabe and Ledeczi research groups from Vanderbilt, the universities of Michigan, Notre Dame, Delaware, Houston and Minnesota, along with Boise and Wayne state universities, to further improve MoSDeF.

Already, the toolkit has been used in published results and ongoing research projects with an impressive 30,000-plus downloads from the Anaconda Cloud software distribution site.
In some fields, the ability to reproduce an experiment and obtain the same results is inherently more difficult—not because the science is unsound but because the details provided in a peer-reviewed publication aren’t enough to recreate the conditions.
The challenges are especially acute in simulating soft matter systems, defined as anything easily deformed at room temperature, such as liquids, polymers, foams, gels and most biological materials. Performing a molecular simulation of such a system involves multiple steps traditionally done one at a time by researchers in a bespoke fashion.

The complexity of soft matter simulations, with hundreds of variables that must be assigned values as the simulation is set up and run, is the source of error and irreproducibility. Distrust of peer-reviewed molecular simulation results is sufficiently high that many groups will repeat a published study to confirm them, tying up computational resources and researcher time.
“I am pretty sure every researcher in our field has had the experience of going down the rabbit hole of trying to confirm a previously published study, only to find in the end that one of these hundreds of unpublished variables was assigned incorrectly,” said Cummings, a globally recognized leader in molecular theory and simulation.
MoSDeF dramatically reduces this problem by automating as many steps as possible.
Consider one key aspect of a molecular simulation: the forcefield, which is a mathematical model for how molecules interact with each other. For a complex molecule, the forcefield can easily have 100 parameters. If a system is a mixture of four or five such molecules, the number of parameters skyrockets. These parameters are made available when the forcefield is first published, but often the original publication will have typographical errors or mistakes in units that are corrected in subsequent papers, or the parameters are further optimized.
One component of MoSDeF provides validated parameters and applies forcefields automatically.
“I like to think of an analogy to manufacturing,” Cummings said. “If an artisan potter makes coffee mugs, each will be slightly different. But in an automated manufacturing environment, they will be replicas of each other. Additionally, if someone sets up the same factory with the same equipment in another location, the same coffee mugs will be produced—that is the essence of reproducibility.”
To have a broad impact, all modules and workflows developed for MoSDeF build on the scientific Python stack to enable transparency and ease entry for new users. The Python packages simplify the creation, atom-typing and simulation of complex molecular models.
“By using freely available tools designed for collaborative code development, such as GitHub and Slack, we are creating a community-developed effort,” said McCabe, Cornelius Vanderbilt Professor of Engineering.
For more information, visit mosdef.org.
MoSDeF is supported by the National Science Foundation under Grant No. 18358741, 1047828 and 1535150
Contact: Brenda Ellis, 615 343-6314
brenda.ellis@vanderbilt.edu